
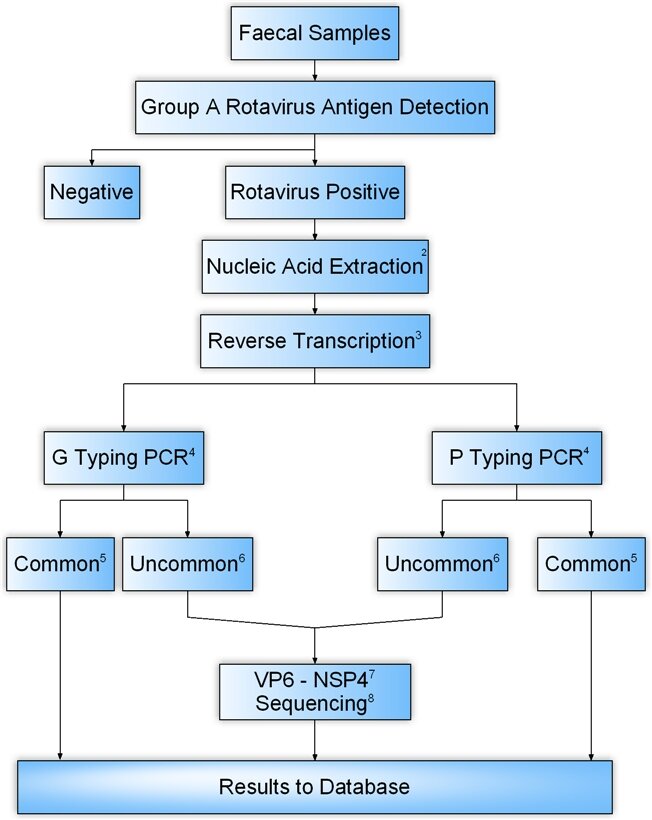
1This algorithm relies on the characterisation of strains using molecular methods and provide comprehensive typing data. Its use allows not only the detection of unusual or uncommon strains, but also G and P combinations suggesting reassortment and/or zoonotic transmission. The methods described here are more labour intensive than serological methods and require specialised facilities, but these would be required as an adjunct to any algorithm which include the use of serotyping methods in order to characterise serologically non-reactive strains.
2The guanidinium isothiocyanate/silica method is recommended for extraction of dsRNA from faecal material (Boom, et al, 1990).
3Reverse transcription with random hexamers will provide cDNA which can be used in both G and P typing PCRs and in PCRs specific for other genes.
4The methods of Gouvea et al [1989], Gentsch, et al [1990] and modified by Iturriza et al [1999; 2000; 2001; 2004 ] should be used.
5Common genotypes will be defined as those included in the vaccine formulation: G1, G2, G3 and G4 and their commonly associated P genotypes: P[4] and P[8].
6Uncommon genotypes will be defined as those not included in the vaccine or common G types in combination with uncommon P types.
7Subgroup and NSP4 genotype should be determined for those strains suspected to have been transmitted from animals to humans as subgroup and NSP4 genotypes segregate tightly according to the species of origin.
8Rotavirus-positive samples of unusual genotypes may provide PCR amplicons in the consensus 1st round PCRs in both or either VP7 and VP4-specific assays but fail to be identified after 2nd round PCRs with type-specific oligonucleotide primers. These strains will be characterised through sequencing of the 1st round PCR products. Direct sequencing of PCR amplicons should be performed and the sequence data, after analysis, should be submitted to the Network database.
Back